Filtering Poor Quality Probes¶
Note: Separate from any filtering that methylcheck does, the qualityMask step of the methylprep processing pipeline excludes a list of probes that are historically poor quality (SeSAMe masks the same list of probes, which are from the Zhou 2016 paper [linked below]). This can be turned off in run_pipeline by specifying quality_mask=False if desired. There are several critera for exclusion of probes that methylcheck offers. These are designed to be in line with past
research that has identified “sketchy” probes. Areas that have polymorphisms, cross-hybridization, repeat sequence elements, or base color changes can affect probe quality. methylcheck’s list_problem_probes() function returns a list of probes excluded based on literature. For each array type, the publication list is as follows:
- 450k: ‘Chen2013,’ ‘Price2013’, ‘Zhou2016’, ‘Naeem2014’, ‘DacaRoszak2015’
- EPIC: ‘Zhou2016’, ‘McCartney2016’
The articles are linked above, for any users that would like more detail on what probes are considered problematic and what criteria the authors have used to identify them. The lists are also formatted in the acceptable input style for the list_problem_probes function. See below for examples of its usage.
[1]:
import methylcheck
from pathlib import Path
filepath = Path('/Users/patriciagirardi/tutorial/GPL21145')
Available probe exclusion lists¶
We’ve imported lists of methylation probes that various researchers have previously deemed to be “sketchy.” You can use methylcheck to remove these probes – by referring to the list by the publication’s first author name, or by the reason these probes should be excluded from analysis.
[2]:
# this code will print the criteria reason (either a publication or a type of issue, like Polymorphism)
# as well as the number of probes excluded for that reason
criteria = ['Chen2013', 'Price2013', 'Naeem2014', 'DacaRoszak2015','Polymorphism',
'CrossHybridization', 'BaseColorChange', 'RepeatSequenceElements']
EPIC_criteria = ['McCartney2016', 'Zhou2016', 'Polymorphism', 'CrossHybridization', 'BaseColorChange', 'RepeatSequenceElements']
print('450k probe exclusion criteria and number of probes excluded:')
for crit in criteria:
print(crit, '--', len(methylcheck.list_problem_probes('450k', [crit])))
print('\nEPIC probe exclusion criteria and number of probes excluded:')
for crit in EPIC_criteria:
print(crit, '--', len(methylcheck.list_problem_probes('EPIC', [crit])))
450k probe exclusion criteria and number of probes excluded:
Chen2013 -- 265410
Price2013 -- 213246
Naeem2014 -- 128695
DacaRoszak2015 -- 89678
Polymorphism -- 289952
CrossHybridization -- 92524
BaseColorChange -- 359
RepeatSequenceElements -- 96631
EPIC probe exclusion criteria and number of probes excluded:
McCartney2016 -- 326267
Zhou2016 -- 178671
Polymorphism -- 346033
CrossHybridization -- 108172
BaseColorChange -- 406
RepeatSequenceElements -- 0
[3]:
# users may also get the list of probe names that are excluded for any of the criteria
methylcheck.list_problem_probes(array='epic', criteria=['Polymorphism'])[0:5]
[3]:
['cg14670079', 'cg26778521', 'cg04785903', 'cg15989966', 'cg21932343']
Filtering poor quality probes¶
[4]:
# leave criteria undefined to list all problem probes for that array type
sketchy_probes_list = methylcheck.list_problem_probes(array='epic')
[5]:
df = methylcheck.load(filepath)
# with the sketchy_probes_list defined above, we can use methylcheck.exclude_probes() to remove all the unwanted probes
excluded_df = methylcheck.exclude_probes(df, sketchy_probes_list)
excluded_df.head()
Files: 100%|██████████| 1/1 [00:00<00:00, 4.75it/s]
INFO:methylcheck.load_processed:loaded data (865859, 16) from 1 pickled files (0.253s)
Of {len(df.index)} probes, {post_overlap} matched, yielding {len(df.index)-post_overlap} probes after filtering.
[5]:
| 203163220027_R01C01 | 203163220027_R02C01 | 203163220027_R03C01 | 203163220027_R04C01 | 203163220027_R05C01 | 203163220027_R06C01 | 203163220027_R07C01 | 203163220027_R08C01 | 203175700025_R01C01 | 203175700025_R02C01 | 203175700025_R03C01 | 203175700025_R04C01 | 203175700025_R05C01 | 203175700025_R06C01 | 203175700025_R07C01 | 203175700025_R08C01 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IlmnID | ||||||||||||||||
| cg00000029 | 0.852 | 0.749 | 0.739 | NaN | 0.891 | 0.896 | 0.757 | 0.734 | 0.806 | 0.881 | 0.740 | 0.885 | 0.693 | 0.779 | 0.617 | 0.891 |
| cg00000109 | 0.943 | 0.916 | 0.884 | 0.951 | 0.936 | 0.774 | 0.932 | 0.920 | 0.898 | 0.938 | 0.931 | 0.953 | 0.899 | 0.889 | 0.734 | 0.892 |
| cg00000165 | 0.237 | 0.733 | 0.374 | 0.639 | 0.397 | 0.564 | 0.486 | 0.194 | 0.163 | 0.557 | 0.531 | 0.856 | 0.171 | 0.173 | 0.865 | 0.581 |
| cg00000221 | 0.943 | 0.937 | 0.939 | 0.926 | 0.942 | 0.945 | 0.938 | 0.933 | 0.949 | 0.936 | 0.934 | 0.949 | 0.944 | 0.937 | 0.942 | 0.934 |
| cg00000236 | 0.922 | 0.934 | 0.919 | 0.881 | 0.926 | 0.882 | 0.927 | 0.774 | 0.937 | 0.957 | 0.932 | 0.952 | 0.923 | 0.938 | 0.932 | 0.951 |
[6]:
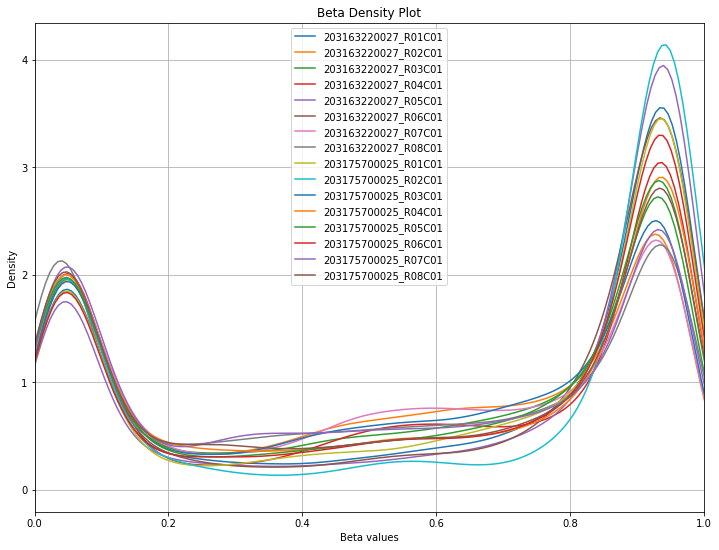
methylcheck.beta_density_plot(excluded_df)
WARNING:methylcheck.samples.postprocessQC:Your data contains 6103 missing probe values per sample, (97656 overall). For a list per sample, use verbose=True
INFO:numexpr.utils:NumExpr defaulting to 8 threads.
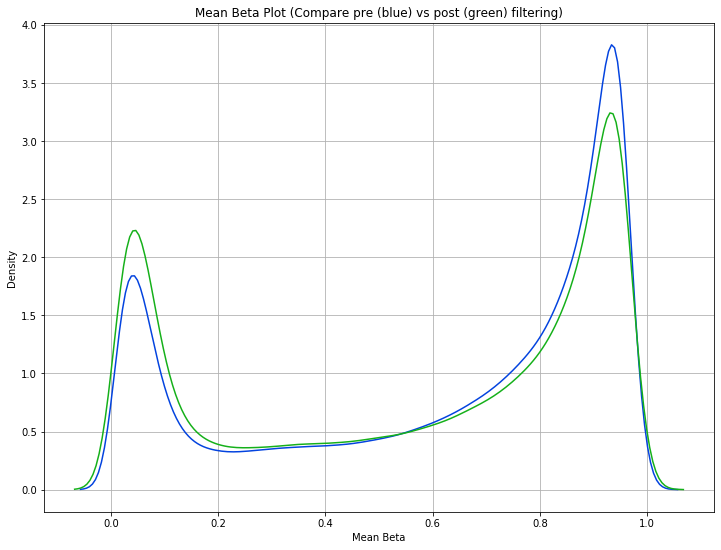
To get a sense for how this affects the data, users may want to compare the mean beta density distribution before and after probe filtering.
[7]:
methylcheck.mean_beta_compare(df, excluded_df)
Filtering sex-linked probes and control probes¶
Other probe types that are often filtered out are sex-linked probes and quality control probes used by Illumina. Quality control probes are automatically filtered out in methylprep processing with the default qualityMask, so there’s no need to run exclude_sex_control_probes if you processed your data with methylprep. Otherwise, we recommend methylcheck’s exclude_sex_control_probes to remove both sex-linked probes and quality control probes.
[8]:
filtered_df = methylcheck.exclude_sex_control_probes(excluded_df, 'epic', no_sex=True, no_control=True, verbose=True)
epic: Removed 12360 sex-linked probes from 16 samples. 464871 probes remaining.
[9]:
filtered_df.head()
[9]:
| 203163220027_R01C01 | 203163220027_R02C01 | 203163220027_R03C01 | 203163220027_R04C01 | 203163220027_R05C01 | 203163220027_R06C01 | 203163220027_R07C01 | 203163220027_R08C01 | 203175700025_R01C01 | 203175700025_R02C01 | 203175700025_R03C01 | 203175700025_R04C01 | 203175700025_R05C01 | 203175700025_R06C01 | 203175700025_R07C01 | 203175700025_R08C01 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IlmnID | ||||||||||||||||
| cg00000029 | 0.852 | 0.749 | 0.739 | NaN | 0.891 | 0.896 | 0.757 | 0.734 | 0.806 | 0.881 | 0.740 | 0.885 | 0.693 | 0.779 | 0.617 | 0.891 |
| cg00000109 | 0.943 | 0.916 | 0.884 | 0.951 | 0.936 | 0.774 | 0.932 | 0.920 | 0.898 | 0.938 | 0.931 | 0.953 | 0.899 | 0.889 | 0.734 | 0.892 |
| cg00000165 | 0.237 | 0.733 | 0.374 | 0.639 | 0.397 | 0.564 | 0.486 | 0.194 | 0.163 | 0.557 | 0.531 | 0.856 | 0.171 | 0.173 | 0.865 | 0.581 |
| cg00000221 | 0.943 | 0.937 | 0.939 | 0.926 | 0.942 | 0.945 | 0.938 | 0.933 | 0.949 | 0.936 | 0.934 | 0.949 | 0.944 | 0.937 | 0.942 | 0.934 |
| cg00000236 | 0.922 | 0.934 | 0.919 | 0.881 | 0.926 | 0.882 | 0.927 | 0.774 | 0.937 | 0.957 | 0.932 | 0.952 | 0.923 | 0.938 | 0.932 | 0.951 |
[10]:
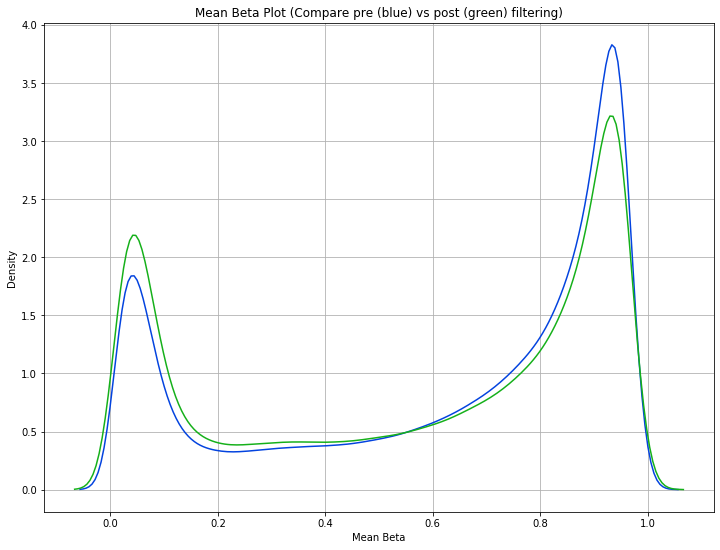
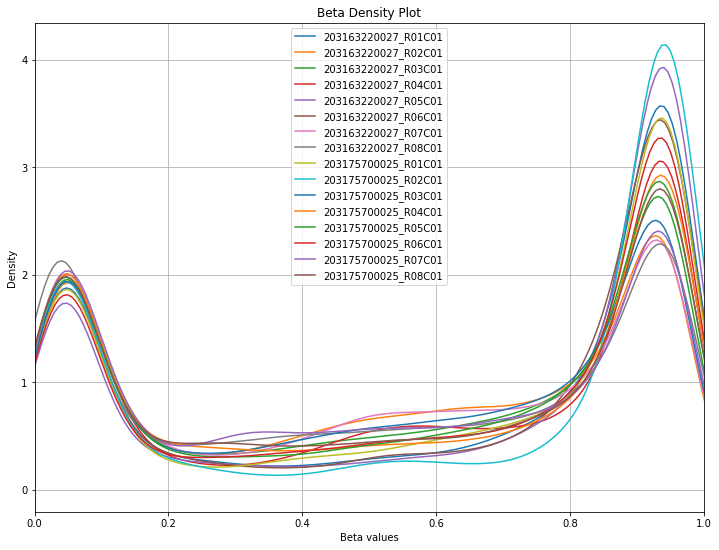
methylcheck.beta_density_plot(filtered_df)
WARNING:methylcheck.samples.postprocessQC:Your data contains 5626 missing probe values per sample, (90031 overall). For a list per sample, use verbose=True
[11]:
methylcheck.mean_beta_compare(df, filtered_df)